The last 20 years of angiogenesis research have been dominated by molecular biology. The identification of the molecular players started with the discovery of VEGF in 1989 (Keck et al. 1989; Leung et al. 1989; Plouët et al. 1989) and the discovery of VEGF-C in 1996 (Joukov et al. 1996; Lee et al. 1996). Several large scale sequencing projects have contributed to the identification of genes involved in angiogenesis, and the focus of today's research is to understand rather than to discover. The task of understanding seems tremendous, despite major technological advances such as genomics and proteomics.
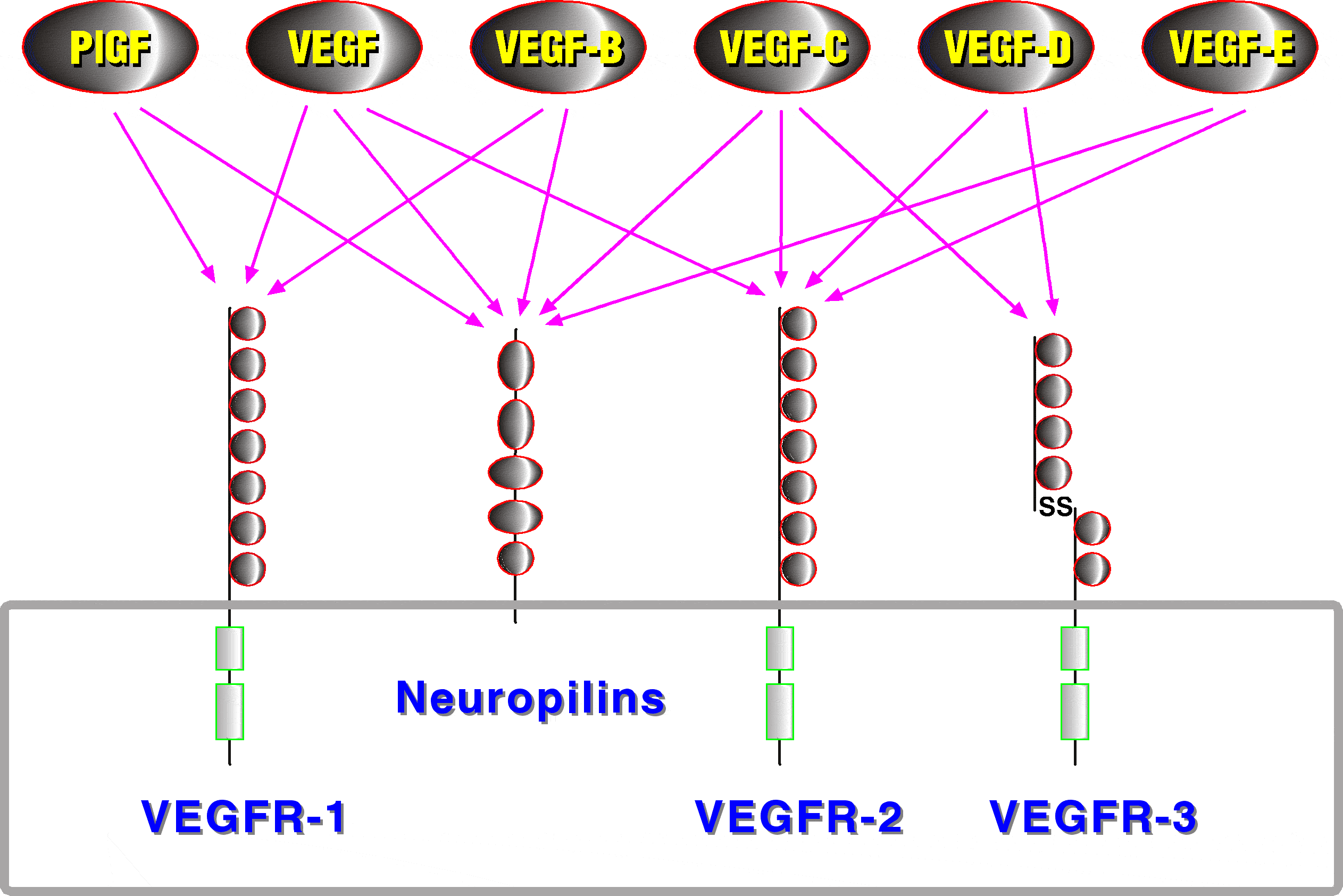
Polypeptide growth factors and their receptors are major components of the regulatory machinery governing angiogenesis. Central positions in this machinery are occupied by the VEGF receptor tyrosine kinases. VEGF receptors are largely specific for endothelial cells. In mammals three VEGF receptors interact with five different VEGFs, each of which has a characteristic receptor-binding pattern (Figure 2). However, other growth factor and receptor families provide major contributions to vessel differentiation, notably the Tie receptors with their angiopoietin ligands (reviewed by Jones et al. 2001), the ephrins and their receptors (reviewed by Cheng et al. 2002), the neuropilins, which can act as co-receptors for several VEGF family members (reviewed by Neufeld et al. 2002) and the PDGFs, which do not act directly on endothelial cells, but instead recruit pericytes and smooth muscle cells to coat the endothelial tube, which is indispensible for vessel stabilization (reviewed by Betsholtz et al. 2001).
The "par excellence" key molecule of vascular development is vascular endothelial growth factor (VEGF, also called VEGF-A to distinguish it from the other VEGF family members). The function of VEGF in vasculogenesis and angiogenesis has been extensively analyzed and reviewed (e.g. Neufeld et al. 1999; Ferrara 2001). VEGF binds to VEGFR-1 and VEGFR-2. VEGFR-2 seems to mediate most, if not all, of the biological effects of VEGF, while VEGFR-1 has probably a modifying function, mostly by acting as a decoy receptor. In VEGFR-2 knockout mice both hematopoiesis and vasculogenesis are blocked (Shalaby et al. 1995), whereas in VEGFR-1 knockout mice the remodeling of the primary vascular plexus fails due to increased hemangioblast commitment of mesenchymal precursors (Fong et al. 1995; Fong et al. 1999). Mice in which the kinase domain of VEGFR-1 is deleted appeared normal, arguing strongly for a primarily non-signaling function of VEGFR-1 (Hiratsuka et al. 1998). Additionally, the viral VEGF homologue VEGF-E, which does not bind VEGFR-1, is able to carry out most of the functions that VEGF does (Meyer et al. 1999; Wise et al. 1999). In mouse development, VEGF expression starts at E 4.0 (Miquerol et al. 1999) before the onset of vasculogenesis. Activation of VEGFR-2 by VEGF has been both implicated in the migration (Shalaby et al. 1997) and the induction (Bautch et al. 2000) of the endothelial precursor cells. Targeted inactivation of even a single VEGF allele results in embryonic lethality, indicating that tightly regulated VEGF levels are a prerequisite for embryonic development (Carmeliet et al. 1996; Ferrara et al. 1996).
VEGF is induced by hypoxia, and although large vessels are genetically determined, at least the vascularization of some embryonic tissues seems to be directed by hypoxia (Stone et al. 1995). In addition to proliferation, VEGF stimulates endothelial cell migration and vessel permeability (Dvorak et al. 1995). Although endothelial cells are the primary target, VEGF also elicits responses in cells of the monocyte/macrophage hematopoietic lineage, including monocytes (Clauss et al. 1990; Barleon et al. 1996), dendritic cells (Gabrilovich et al. 1996) and osteoclasts (Midy and Plouet 1994; Gerber et al. 1999).
In addition to the predominant VEGF165 species, five other isoforms are generated in humans by alternative splicing: VEGF121, VEGF145, VEGF183, VEGF189, and VEGF206 (Leung et al. 1989; Houck et al. 1991; Tischer et al. 1991; Poltorak et al. 1997; Lei et al. 1998). The isoforms vary in their expression pattern and their binding properties for heparan sulfate proteoglycans, extracellular matrix and co-receptors neuropilin-1 and -2. VEGF121 binds neither heparin nor neuropilin, while VEGF145 and VEGF165 do (Soker et al. 1996; Gluzman-Poltorak et al. 2000). The interaction with neuropilins might explain the differential biological potency, despite having an equal affinity towards VEGF receptors. For cultured endothelial cells VEGF121 is substantially less mitogenic than VEGF165 (Keyt et al. 1996a), and in new-born gene targeted mice VEGF120 cannot compensate for the loss of the longer isoforms, leading to ischemic cardiomyopathy and death (Carmeliet et al. 1999b). The ability to interact with glycosaminoglycans also prolongs the biological half-life of VEGF165 compared to VEGF121, since glypican-1 can act as an extracellular "chaperone" for VEGF (Gengrinovitch et al. 1999).
Figure 2. VEGF family members and their receptors
VEGF and PDGFs are members of the cystine knot growth factor superfamily (McDonald and Hendrickson 1993). PDGF-B was the first member of the PDGF/VEGF family whose structure was solved by X-ray crystallography, followed by VEGF (both alone and complexed to domain 2 of VEGFR-1; Oefner et al. 1992; Muller et al. 1997b; Wiesmann et al. 1997) and PlGF (Iyer et al. 2001). These structures describe the receptor-binding domain, which is largely identical with the VEGF homology domain. Besides the VEGF homology domain, most PDGF/VEGF family members contain at least one additional domain with unique characteristics. In case of VEGF this additional domain can fold independently from the VEGF homology domain, and its structure was determined by NMR spectroscopy (Fairbrother et al. 1998).
VEGF is an antiparallel dimer, covalently linked by two disulfide bridges between Cys-51 and Cys-60. Its cystine knot is located lateral to a central 4-stranded antiparallel
![]() -sheet. One VEGF molecule ligates two receptor molecules by virtue of its two receptor-binding sites, which lie at both ends of the flat and elongated molecule (see
Figure 4A). Both subunits contribute to each individual binding site in a 2 to 1 surface area ratio (Muller et al. 1997a;
Muller et al. 1997b;
Wiesmann et al. 1997). There is no conclusive data about the necessity of disulfide bridges for structural integrity and thermodynamic stability of cystine-knot growth factors. Dimerization of VEGF-C, VEGF-D and NGF does not utilize intersubunit disulfide bridges (McDonald et al. 1991;
Joukov et al. 1996). Intersubunit disulfide bridges are dispensable for the integrity of PDGF-BB (Kenney et al. 1994). The integrity of VEGF produced by mammalian cells was compromised when the intersubunit bridge-forming cysteines were mutated (Potgens et al. 1994). However, VEGF mutants produced in E.coli, that lacked either inter- or intrasubunit cysteine bridges could be folded in vitro, but these mutants were not tested for their biological activity (Heiring and Muller 2001).
-sheet. One VEGF molecule ligates two receptor molecules by virtue of its two receptor-binding sites, which lie at both ends of the flat and elongated molecule (see
Figure 4A). Both subunits contribute to each individual binding site in a 2 to 1 surface area ratio (Muller et al. 1997a;
Muller et al. 1997b;
Wiesmann et al. 1997). There is no conclusive data about the necessity of disulfide bridges for structural integrity and thermodynamic stability of cystine-knot growth factors. Dimerization of VEGF-C, VEGF-D and NGF does not utilize intersubunit disulfide bridges (McDonald et al. 1991;
Joukov et al. 1996). Intersubunit disulfide bridges are dispensable for the integrity of PDGF-BB (Kenney et al. 1994). The integrity of VEGF produced by mammalian cells was compromised when the intersubunit bridge-forming cysteines were mutated (Potgens et al. 1994). However, VEGF mutants produced in E.coli, that lacked either inter- or intrasubunit cysteine bridges could be folded in vitro, but these mutants were not tested for their biological activity (Heiring and Muller 2001).
Both VEGF121 and VEGF-C contain an additional cysteine residue in or close to the VEGF homology domain, which in case of VEGF121 has been shown to be capable of forming an additional interchain disulfide bond (Keck et al. 1997).
Human VEGF receptors are glycosylated type I integral membrane proteins. Seven immunoglobulin-like domains form the extracellular portion and a split kinase domain forms most of its intracellular part. VEGF receptors are close relatives of the PDGF receptors. Domains 6 and 7 of VEGF receptors are thought to be a result of a duplication event in a putative common ancestor gene of the VEGF receptors, PDGF receptors, the CSF-1 receptor (c-Fms) and the SCF receptor (c-Kit). Thus immunoglobulin-like domains 1 to 5 of VEGF receptors correspond to the whole extracellular domain of PDGF receptors, c-Fms and c-Kit (Claesson-Welsh et al. 1988, 1989; Shibuya et al. 1990).
Figure 3. Alignment of selected VEGFs| GREY CHARACTERS | - signal peptide, | GREY BACKGROUND | - BR3P motif, | BLACK BACKGROUND | - putative N-glycosylation sites; |
| the VHD is boxed and the 8 conserved cysteines are indicated by asterisks (*). | |||||

The second Ig-like domain of VEGFR-1 (VEGFR-1D2) was shown to be sufficient for VEGF binding (Wiesmann et al. 1997), contradicting earlier studies (Davis-Smyth et al. 1996;
Barleon et al. 1997;
Cunningham et al. 1997b). The crystal structure rationalized this discrepancy, showing that earlier domain deletions cut into the last
![]() -strand of domain 2 or had extra residues that were likely to destabilize the overall folding (Wiesmann et al. 1997). Deletion constructs containing only domains 2 and 3 of both VEGFR-1 and VEGFR-2 bind VEGF with nearly wild type affinities. In VEGFR-1 further deletion of domain 3 decreases the affinity for VEGF only 20-fold, while the corresponding deletion in VEGFR-2 results in a 1000-fold decrease, showing that the relative importance of domain 3 differs between VEGFR-1 and VEGFR-2 (Wiesmann et al. 1997;
Fuh et al. 1998).
-strand of domain 2 or had extra residues that were likely to destabilize the overall folding (Wiesmann et al. 1997). Deletion constructs containing only domains 2 and 3 of both VEGFR-1 and VEGFR-2 bind VEGF with nearly wild type affinities. In VEGFR-1 further deletion of domain 3 decreases the affinity for VEGF only 20-fold, while the corresponding deletion in VEGFR-2 results in a 1000-fold decrease, showing that the relative importance of domain 3 differs between VEGFR-1 and VEGFR-2 (Wiesmann et al. 1997;
Fuh et al. 1998).
Despite several non-canonical features VEGFR-1D2
is a member of the I-set of the immunoglobulin superfamily (Harpaz and Chothia 1994;
Wiesmann et al. 1997). The sequence deviations in VEGFR-1D2
from I-set positions are conserved in the second domain of other closely related receptors such as VEGFR-2, VEGFR-3, PDGFR![]() , PDGFR
, PDGFR![]() , c-fms and c-kit (Wiesmann et al. 1997). These related receptors also bind their ligand (at least partly) with their second domains (Blechman et al. 1995;
Mahadevan et al. 1995;
Lemmon et al. 1997;
Fuh et al. 1998). Similarly to FGFs, heparin or heparan sulfate proteoglycans have been shown to be essential for high-affinity binding of VEGF to VEGFR-2 (Gitay-Goren et al. 1992). In VEGFR-2 the heparin-binding domain does not map anywhere near the ligand-binding domain (Dougher et al. 1997), while in the FGF2/FGF receptor-1/heparin complex two heparin molecules are integral components of the receptor-ligand interface (Schlessinger et al. 2000).
, c-fms and c-kit (Wiesmann et al. 1997). These related receptors also bind their ligand (at least partly) with their second domains (Blechman et al. 1995;
Mahadevan et al. 1995;
Lemmon et al. 1997;
Fuh et al. 1998). Similarly to FGFs, heparin or heparan sulfate proteoglycans have been shown to be essential for high-affinity binding of VEGF to VEGFR-2 (Gitay-Goren et al. 1992). In VEGFR-2 the heparin-binding domain does not map anywhere near the ligand-binding domain (Dougher et al. 1997), while in the FGF2/FGF receptor-1/heparin complex two heparin molecules are integral components of the receptor-ligand interface (Schlessinger et al. 2000).
Mutagenesis of VEGF had suggested three negatively charged key residues for the interaction with VEGFR-1 (Asp-63, Glu-64 and Glu-67; Keyt et al. 1996b), but the co-crystallization of VEGF with VEGFR-1D2 showed, that only Asp-63 participated in receptor binding via a charge-mediated interaction with Arg-224 of VEGFR-1D2. The VEGFR-1 binding interface is dominated by hydrophobic contacts and is surprisingly flat, showing no predominant knob-into-hole interactions (Wiesmann et al. 1997). VEGFR-1D2 does not undergo any significant conformational changes upon ligand binding (Starovasnik et al. 1999), but the flexible loops that surround the rigid core of VEGF seem to be necessary for achieving promiscuity in binding two different receptors (Muller et al. 1997a).
The VEGFR-2 binding interface of VEGF was identified by alanine-scanning mutagenesis (Muller et al. 1997b). Seven mostly hydrophobic residues were found to be significant for strong VEGFR-2 binding, five of which are localized in the VEGFR-1 binding interface of VEGF (Phe-17, Ile-46, Gln-79, Ile-83 and Pro-85), suggesting that the binding sites for VEGFR-1 and VEGFR-2 are very similar. The two remaining residues are located outside the VEGFR-1D2 binding interface (Ile-43 and Glu-64), but may be part of the binding interface in the native complex (Wiesmann et al. 1997). Previous studies had identified Arg-82, Lys-84 and His-86 as the most important residues for KDR binding ( Keyt et al. 1996b), however this mutagenesis study was not systematic and did not include 6 of the 7 amino acids that Muller et al. identified later in a systematic approach (Muller et al. 1997b; Wiesmann et al. 1997).
Predimerized VEGFR-2 binds VEGF about 100-fold more tightly than monomeric VEGFR-2, while predimerized VEGFR-1 binds VEGF only 2-fold more tightly than monomeric VEGFR-1 (Wiesmann et al. 1997; Fuh et al. 1998). Receptor-receptor interactions might be more prominent in VEGFR-1 than in VEGFR-2, especially under the assay conditions that did not include heparin, which is necessary for the receptor-receptor interaction of VEGFR-2 (Dougher et al. 1997).
The C-terminal domain of VEGF165 (also called VEGF55) constitutes a heparin-binding domain similar to the one of plasminogen, but unrelated to the FGF type heparin binding domain (Fairbrother et al. 1998). Based on the homology of the VEGF heparin-binding domain to the C-terminal tail of VEGF-C and -D, one could expect VEGF-C to bind heparin, although such binding has not been reported in the literature.
Placenta growth factor (PlGF) and VEGF-B differ from VEGF in that they do not bind VEGFR-2 (Park et al. 1994; Olofsson et al. 1998). Both PlGF and VEGF-B exist in two different isoforms (PlGF-1 and PlGF-2; VEGF-B167 and VEGF-B186), which are generated by differential mRNA splicing (Maglione et al. 1993; Olofsson et al. 1996).
Figure 4. Structure of VEGFs: Uncoupling receptor binding and specificity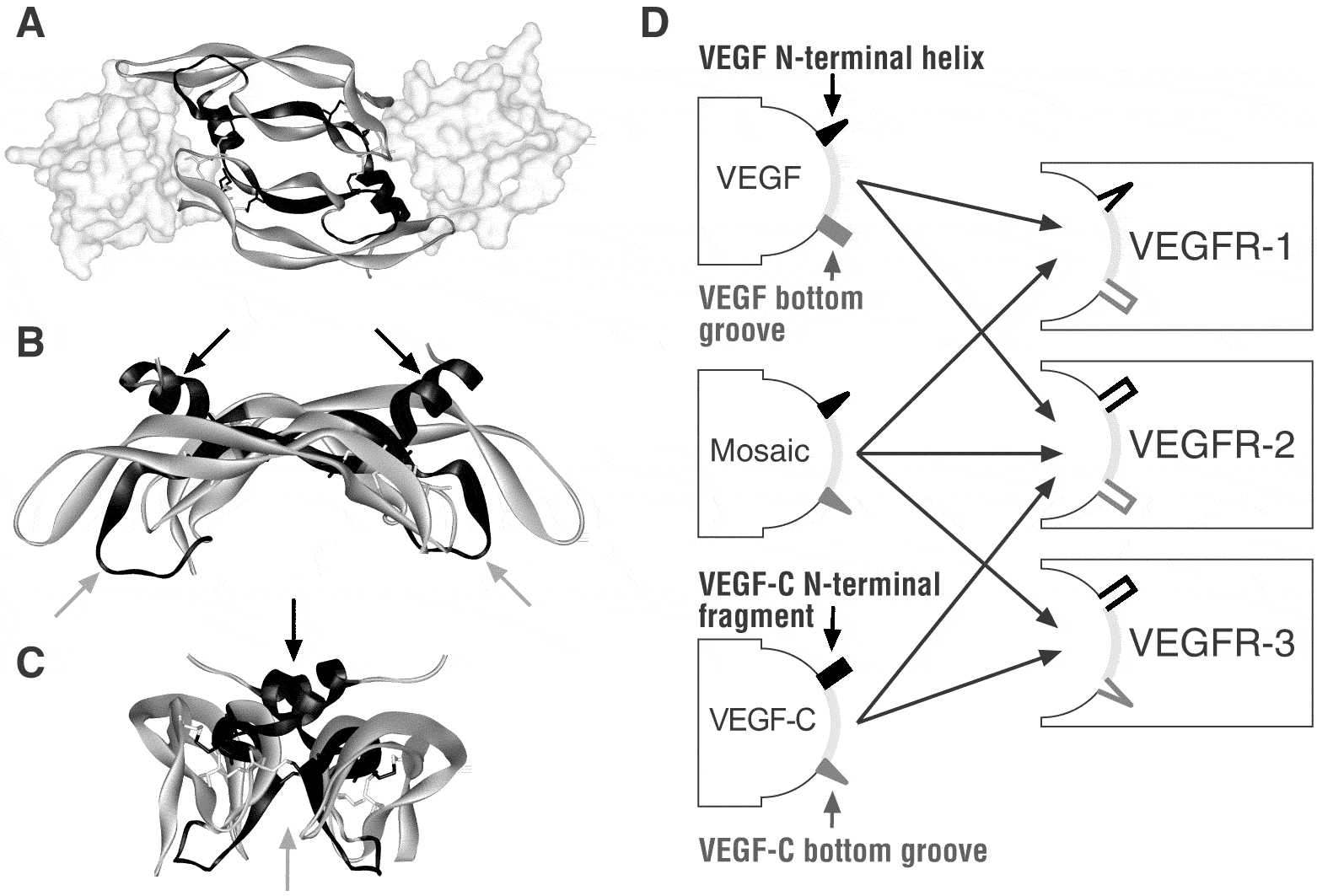
While alternative splicing of VEGF and PlGF mRNA successively integrates additional in-frame basic stretches into the polypeptide, the alternative splicing of VEGF-B mRNA results in a frame shift and two non-homologous C-termini. The C-terminus of VEGF-B167 is similar to VEGF55, whereas VEGF-B186 has a unique hydrophobic C-terminus, which can be for the most part removed by proteolytic processing at Arg-127. This cleavage regulates the affinity of VEGF-B186 for its co-receptor neuropilin-1, which also binds VEGF-B167 and PlGF-2 (Migdal et al. 1998; Makinen et al. 1999). Unlike all other VEGF family members, VEGF-B does not contain any N-linked carbohydrates: VEGF-B167 is not glycosylated at all and VEGF-B186 becomes O-glycosylated (Olofsson et al. 1996).
VEGF-B has a wide tissue distribution that overlaps substantially with that of VEGF. In adults it is most abundant in heart, skeletal muscle and brown fat. VEGF-B167 seems to be the predominant isoform under physiological conditions (Olofsson et al. 1996; Lagercrantz et al. 1998; Aase et al. 1999; Li et al. 2001). The function of PlGF and VEGF-B needs still further characterization. In most systems VEGFR-1 mediated biological responses are absent or weak (II; Waltenberger et al. 1994; Sawano et al. 1996). Both PlGF and VEGF-B knockout mice are viable and fertile, and develop a normal vasculature. Studies in these knockout mice link PlGF specifically with repair angiogenesis (Carmeliet et al. 2001; Luttun et al. 2002) and VEGF-B weakly with cardiac function (Aase et al. 2001).
Some predators make use of VEGF-like molecules with devastating results for their prey: molecules with approximately 50% identity to VEGF are apparently common components of snake venoms. In addition to the induction of hypotensic shock, the increase in vascular permeability presumably facilitates access of the neurotoxic venom components to their target cells (Komori et al. 1999; Gasmi et al. 2000; Junqueira de Azevedo et al. 2001; Gasmi et al. 2002). Another innovative use for VEGF-like molecules can be seen in some parapoxviruses. Orf virus strains NZ2, NZ7 and D1701 have captured a VEGF-like gene from their mammalian hosts, and the vascular lesions caused by these viral VEGFs (collectively known as VEGF-E) are probably instrumental in viral spread (Lyttle et al. 1994; Ogawa et al. 1998; Meyer et al. 1999; Wise et al. 1999; Savory et al. 2000).
A close paralogue of VEGFR-1 and -2 is VEGFR-3. Its expression becomes restricted to lymphatic endothelial cells during embryogenesis (Kaipainen et al. 1995; Kukk et al. 1996). The search for its ligand led to the identification two lymphangiogenic growth factors: VEGF-C and VEGF-D (Joukov et al. 1996; III).
VEGF-C and VEGF-D specifically induce the proliferation of lymphatic endothelial cells in transgenic mice and in the CAM (I; II; Veikkola et al. 2001; own unpublished data). In the same assays VEGF is specific for blood vessels and does not induce any changes in the lymphatic vasculature (II; Detmar et al. 1998). However, several reports confirm that both VEGF-C and VEGF-D can exercise significant angiogenic potency both in vitro and in vivo (Lee et al. 1996; Cao et al. 1998; Witzenbichler et al. 1998; Marconcini et al. 1999; Byzova et al. 2002). Discrepancies concerning the angiogenic versus the lymphangiogenic potential of VEGF-C and VEGF-D are believed to be a result of different extracellular processing (Joukov et al. 1997; Stacker et al. 1999a). This, however, does not explain experimental results showing that also the lymphangiogenic potential of mature VEGF-C exceeds by far its angiogenic potential (II).
Mice deficient in VEGFR-3 died from cardiovascular failure at E9.5 (Dumont et al. 1998) due to an impaired remodeling and maturation of large vessels. VEGFR-3 thus appears to a have an essential role before the formation of the lymphatic system, when VEGFR-3 is still ubiquitously expressed on all endothelial cells. In VEGF knock-out embryos endothelial differentiation is not completely blocked unlike in the VEGFR-2 knock-out mice, suggesting that VEGF-C indeed does activate VEGFR-2 in early embryonic development (Carmeliet et al. 1996; Ferrara et al. 1996; Shalaby et al. 1997). At least in vitro VEGF-C was able to replace VEGF in a differentiation assay using avian hemangioblasts (Eichmann et al. 1997). More insight into the role of VEGFR-3 in lymphatic development could be obtained by a conditional knockout. However, caution was recommended when interpreting mouse VEGFR-3 data, since in humans an endogenous retrovirus disrupts the splice pattern, giving rise to a C-terminally shortened receptor isoform, whose signaling properties differ from the long isoform (Pajusola et al. 1993; Borg et al. 1995; Hughes 2001).
VEGF-C and VEGF-D differ from other VEGF family members by the presence of long N- and C-terminal extensions flanking the VEGF-homology domain (Joukov et al. 1996; Lee et al. 1996; Orlandini et al. 1996; Yamada et al. 1997; III). The N-terminal propeptide does not contain any recognizable motifs, while the C-terminal domain contains a repetitive cysteine pattern (Cys-X10-Cys-X-Cys-X-Cys), homologous to a motif first identified in a silk-like secretory Balbiani ring 3 protein produced by larval salivary glands of the midge Chironomus tentans.
The VEGF homology domain (VHD) of VEGF-C exhibits 35% identity to VEGF. It is encoded by exons 3 and 4 of the seven exons (Chilov et al. 1997), which is a feature conserved in other members of the VEGF family (Tischer et al. 1991; Maglione et al. 1993; Olofsson et al. 1996). The VEGF homology domains of VEGF-C and VEGF-D are 60% identical. VEGF-C is synthesized as a precursor protein, which undergoes subsequent proteolytic processing (Joukov et al. 1996; Joukov et al. 1997). The C-terminal domain is cleaved upon secretion, but remains bound to the N-terminal domain by disulfide bonds, giving rise to a disulfide linked tetramer composed of 29-kDa and 31-kDa polypeptides. Proteolytic processing of the N-terminal propeptide releases the mature form, which consists of two 21-kDa polypeptide chains corresponding largely to the VEGF homology domain (Joukov et al. 1997). The 29/31-kDa form seems to be the most prevalent form of VEGF-C in various biological systems (Lee et al. 1996; Hu et al. 1997; Joukov et al. 1997; Eichmann et al. 1998; Hiltunen et al. 2000). Incomplete and additional proteolytic processing leads to two minor fragments that migrate on reducing SDS-PAGE with an apparent molecular weight of 15 and 43 kDa. The 15-kDa fragment represents the N-terminal propeptide while the 43-kDa form presumably represents its complement (the VHD and the C-terminal domain). VEGF-D is processed in a similar fashion (Stacker et al. 1999a).
Both the full-length and the mature forms of human VEGF-C bind VEGFR-3 with high affinity (Joukov et al. 1997), while high affinity binding to VEGFR-2 requires proteolytic processing. Full length VEGF-C and the 29/31-kDa form bind VEGFR-2 weakly, while the mature form and the 43-kDa form bind efficiently (Joukov et al. 1997; own unpublished data). The receptor binding profile of VEGF-C seems to be conserved among species, but not the one of VEGF-D, as mouse VEGF-D does not bind mouse VEGFR-2 (Baldwin et al. 2001a). Similar to VEGF, glycosylation of VEGF-C is no prerequisite for receptor binding, but is necessary for efficient secretion (own unpublished data).
Neither the structure of VEGFR-3 nor of any of its ligands has been solved. The high homology of VEGF-C and VEGF in the VHD allows for molecular modeling of this domain and together with mutational data offers some limited insights into the nature of the interaction (IV). The primary sequences of VEGF-C and VEGF show major differences in two regions which code for the N-terminal part and for the flexible loop between
![]() -strands 5 and 6. The N-terminal part of the VHD forms an
-strands 5 and 6. The N-terminal part of the VHD forms an
![]() -helix in both VEGF and PlGF (Muller et al. 1997b;
Iyer et al. 2001), but is disordered in PDGF-BB (Oefner et al. 1992) and probably also in VEGF-C. Mutational analysis shows that these two regions are involved in determining receptor specificity in VEGFs (IV). VEGF-C loses its ability to bind and activate VEGFR-2 when Cys-156 is mutated into serine (Joukov et al. 1998). In other members of the PDGF/VEGF family, cysteine residues homologous to this Cys-156 are involved in interchain disulfide bonding, whereas VEGF-C and VEGF-D are noncovalent dimers (Joukov et al. 1997;
Stacker et al. 1999a). This is additional evidence for a role of the bottom groove in receptor binding as previously proposed for VEGF and VEGFR-1 (Wiesmann et al. 1997). Indeed, residues of this region are indispensable for VEGFR-3 binding (IV). Similarly to VEGFR-1 binding, VEGFR-3 binding does not require the presence of the third domain of the receptor (IV). Surprisingly enough, the first domain appeared necessary, although it is unlikely that domain 1 interacts directly with VEGF-C (IV).
-helix in both VEGF and PlGF (Muller et al. 1997b;
Iyer et al. 2001), but is disordered in PDGF-BB (Oefner et al. 1992) and probably also in VEGF-C. Mutational analysis shows that these two regions are involved in determining receptor specificity in VEGFs (IV). VEGF-C loses its ability to bind and activate VEGFR-2 when Cys-156 is mutated into serine (Joukov et al. 1998). In other members of the PDGF/VEGF family, cysteine residues homologous to this Cys-156 are involved in interchain disulfide bonding, whereas VEGF-C and VEGF-D are noncovalent dimers (Joukov et al. 1997;
Stacker et al. 1999a). This is additional evidence for a role of the bottom groove in receptor binding as previously proposed for VEGF and VEGFR-1 (Wiesmann et al. 1997). Indeed, residues of this region are indispensable for VEGFR-3 binding (IV). Similarly to VEGFR-1 binding, VEGFR-3 binding does not require the presence of the third domain of the receptor (IV). Surprisingly enough, the first domain appeared necessary, although it is unlikely that domain 1 interacts directly with VEGF-C (IV).
The existence of mosaic VEGFs that bind all VEGF receptors adds prove to the idea that the binding of VEGFs to their receptors uses a very similar mechanism. The inability of a VEGF to bind to a certain VEGF receptor can be traced to limited specific structural elements (IV). This partial uncoupling of receptor binding determinants from receptor specificity determinants is reminiscent of human growth hormone (HGH) and prolactin. HGH can bind both the HGH receptor and the prolactin receptor, while prolactin binds only to the prolactin receptor (Somers et al. 1994). The C-terminal part of VEGF-C has significant homology to the BR3P protein (Joukov et al. 1996). Also VEGF and VEGF-B167 contain two BR3P repeats in their C-terminal domains (see Figure 3), and the NMR structure of the C-terminal domain of VEGF shows that the two repeats indeed fold into two distinct domains (Fairbrother et al. 1998). Nevertheless, apart from the conserved cysteines, the sequence conservation between the C-terminal domains of VEGF and VEGF-C is quite low.
As expected for paracrine growth factors, expression of the cognate receptors for VEGF-C and VEGF-D is usually found in close proximity to the growth factor-expressing cells. In the mouse, VEGF-C mRNA expression starts around E8.5 in the head mesenchyme and the developing vertebrae. At E12.5, VEGF-C expression is strong in the mesenchyme of the metanephric and jugular area, where the embryonic lymph sacs sprout from the large veins (Kukk et al. 1996). This pattern is conserved between species. In quail and chick embryos VEGF-C was observed in areas that soon after became rich in lymphatic endothelium (Eichmann et al. 1993; Eichmann et al. 1998). In adult mice, the expression of VEGF-C decreases, but its mRNA can still be found in the lung, heart, liver and kidney (Kukk et al. 1996; Fitz et al. 1997; Lymboussaki et al. 1999).
Although VEGF-D and VEGF-C expression overlaps, e.g. in the heart, important differences exist both during development and adult life, notably the high expression of VEGF-D in the lung (Avantaggiato et al. 1998; Farnebo et al. 1999). The regulation of VEGF-C and VEGF-D expression is less well understood than the one of VEGF (Shweiki et al. 1992; Shima et al. 1996; Stein et al. 1998; Oosthuyse et al. 2001). Unlike the VEGF promoter, the VEGF-C promoter lacks hypoxia response elements (Chilov et al. 1997), and thus VEGF-C mRNA levels are not regulated by hypoxia (Enholm et al. 1997). Several growth factors and inflammatory cytokines upregulate VEGF-C expression (Enholm et al. 1997; Ristimaki et al. 1998), whereas steroid hormones act as down-regulators (Laitinen et al. 1997; Ruohola et al. 1999). The VEGF-D promoter has not been characterized extensively, but both hypoxia and cell-cell contacts have been implicated in its regulation (Orlandini and Oliviero 2001; Teng et al. 2002).
The signal transduction pathways of VEGF receptors are still not well understood. Endothelial cells typically express more than one VEGF receptor, making it difficult to assign a specific receptor to any biological effect. On the other hand, non-endothelial cells transfected with a single receptor might lack the endothelial cell-specific signal transduction machinery. Only recently has the use of receptor-specific mutants to activate receptors on isolated primary cultures of endothelial cells improved the situation (Gerber et al. 1998b; Joukov et al. 1998; Makinen et al. 2001b).
It is thought that in endothelial cells the major VEGF-induced mitogenic signal is routed independently from Ras via PLC![]() -PKC to the MAPK cascade (Waltenberger et al. 1994;
Seetharam et al. 1995;
Cunningham et al. 1997a;
Takahashi and Shibuya 1997;
Takahashi et al. 1999). Akt phosphorylation by PI3-K seems to be important for survival signaling (Xia et al. 1996;
Gerber et al. 1998b;
Jiang et al. 2000). This process is dependent on association of VEGFR-2 with vascular endothelial cadherin (Carmeliet et al. 1999a). Akt is also one of the pathways by which VEGF activates the vasorelaxant endothelial nitric oxide synthase (eNOS;
Parenti et al. 1998;
Dimmeler et al. 1999;
Fulton et al. 1999).
-PKC to the MAPK cascade (Waltenberger et al. 1994;
Seetharam et al. 1995;
Cunningham et al. 1997a;
Takahashi and Shibuya 1997;
Takahashi et al. 1999). Akt phosphorylation by PI3-K seems to be important for survival signaling (Xia et al. 1996;
Gerber et al. 1998b;
Jiang et al. 2000). This process is dependent on association of VEGFR-2 with vascular endothelial cadherin (Carmeliet et al. 1999a). Akt is also one of the pathways by which VEGF activates the vasorelaxant endothelial nitric oxide synthase (eNOS;
Parenti et al. 1998;
Dimmeler et al. 1999;
Fulton et al. 1999).
VEGF and integrins potentiate each other in a reciprocal manner. VEGF upgregulates the endothelium-specific ![]() V
V![]() 3
integrin (Senger et al. 1997) and
3
integrin (Senger et al. 1997) and ![]() V
V![]() 3
integrin associates with VEGFR-2 to potentiate VEGF-induced signaling (Soldi et al. 1999). Other target genes of VEGF that have been implicated in various aspects of angiogenesis include many proteases like plasmin and matrix metalloproteinases
(MMPs;
Pepper et al. 1991;
Unemori et al. 1992), cytoskeletal components such as focal adhesion kinase
(FAK;
Abedi and Zachary 1997), anti-apoptotic proteins like Bcl-2 (Gerber et al. 1998a) and transcription factors of the STAT family (Korpelainen et al. 1999).
3
integrin associates with VEGFR-2 to potentiate VEGF-induced signaling (Soldi et al. 1999). Other target genes of VEGF that have been implicated in various aspects of angiogenesis include many proteases like plasmin and matrix metalloproteinases
(MMPs;
Pepper et al. 1991;
Unemori et al. 1992), cytoskeletal components such as focal adhesion kinase
(FAK;
Abedi and Zachary 1997), anti-apoptotic proteins like Bcl-2 (Gerber et al. 1998a) and transcription factors of the STAT family (Korpelainen et al. 1999).
Compared to VEGFR-2, little is known about VEGFR-3-initiated signal transduction. From the two splice variants the short isoform is compromised in its signaling capabilities, presumably due to the absent phosphorylation sites of the C-terminus (Pajusola et al. 1993;
Fournier et al. 1995). When transfected into PAE cells, VEGFR-3-initiated signal transduction appears very similar to VEGFR-2 (Kroll and Waltenberger 1997), Shc becomes tyrosine phosphorylated, and cell proliferation increases (Pajusola et al. 1994;
Fournier et al. 1996;
Wang et al. 1997). Like VEGFR-2, VEGFR-3 associates with Grb2 and PLC
![]() (Pajusola et al. 1994;
Fournier et al. 1995;
Fournier et al. 1996). Also the biological response is similar in PAE cells: migration, actin reorganization, and proliferation (Joukov et al. 1996;
Cao et al. 1998). Only recently have natively VEGFR-3 expressing isolated lymphatic endothelial cells been used to study signal transduction. These studies show distinct differences between VEGFR-2 and VEGFR-3 mediated signaling in the same cell type. E.g. in VEGFR-2 signaling PKC activates p38 MAPK, while in VEGFR-3 signaling p42/p44 MAPK is the target (Gerber et al. 1998b;
Taipale et al. 1999;
Makinen et al. 2001b).
(Pajusola et al. 1994;
Fournier et al. 1995;
Fournier et al. 1996). Also the biological response is similar in PAE cells: migration, actin reorganization, and proliferation (Joukov et al. 1996;
Cao et al. 1998). Only recently have natively VEGFR-3 expressing isolated lymphatic endothelial cells been used to study signal transduction. These studies show distinct differences between VEGFR-2 and VEGFR-3 mediated signaling in the same cell type. E.g. in VEGFR-2 signaling PKC activates p38 MAPK, while in VEGFR-3 signaling p42/p44 MAPK is the target (Gerber et al. 1998b;
Taipale et al. 1999;
Makinen et al. 2001b).